题目:Integrative detection and analysis of structural variation in cancer genomes
期刊:Nature genetics
影响因子:27.125
研究背景
结构变异(SVs),包括倒置、删除、复制阳离子和易位,是大多数癌症基因组的标志。复发性SVs的发现及其对基因组织和表达的分子效应促进我们对肿瘤发生的认识。许多致癌基因已被确认为复发易位的产物,并为药物治疗特别是造血恶性肿瘤提供了成功的靶点。尽管它很重要,但在癌症基因组中鉴定SVs仍然具有挑战性。在这里,作者利用高通量染色体构象捕获(Hi-C)、集成光学映射和全基因组测序,系统地检测正常或癌症样本中的SVs,意图探究癌基因组中,SVs对突变驱动因素的影响。
研究内容及结果
1. 检测肿瘤基因组SV的方法
为了评估对SVs不同检测方法的能力,作者选取了8个癌细胞系和1个典型正常对照(GM12878)(见表1),对它们的WGS、光学测图和Hi-C数据进行比较(图1a),发现三种方法均检测到Caki2细胞中染色体2和3的易位(图1b),通过观察同一区域DNA复制时间谱的显著变化,也证实了这种易位。同时观察到,与正常细胞相比,癌症基因组显示出更多的重组事件,如图1c所示环状基因组结构剖面。
表1肿瘤和正常细胞系的高置信度的SVs数
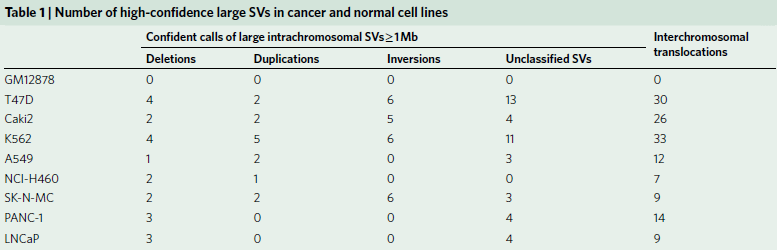
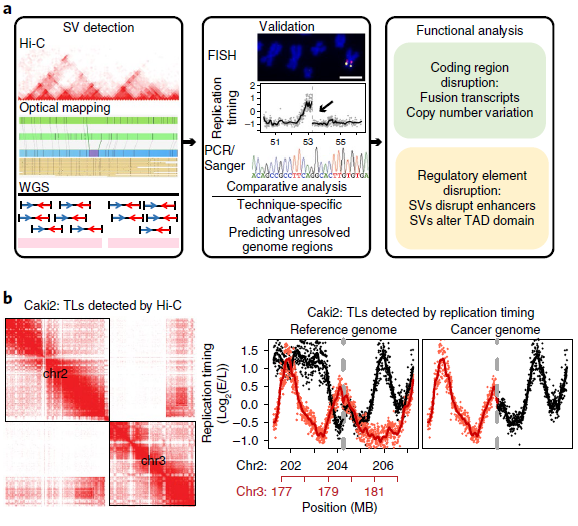
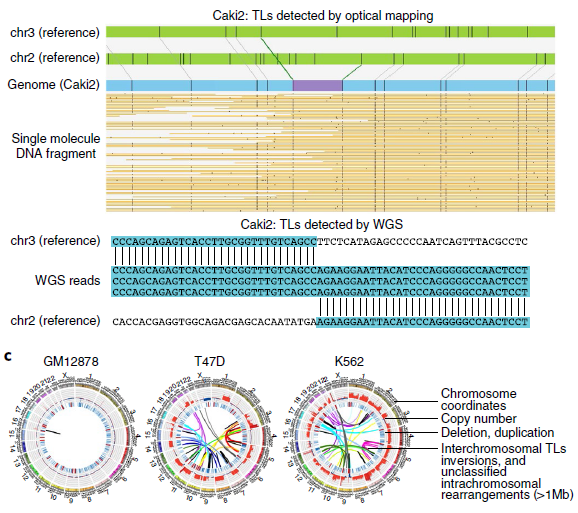
图1 肿瘤基因组SV检测的总体策略
2. 利用Hi-C数据检测大规模重排
在Hi-C实验中,正常细胞染色体间相互作用非常罕见(图2a左)。然而,这这种情况在癌细胞中却相反。例如Caki2癌症在细胞中,观察到了强烈的染色体间相互作用(图2a右),这可能是由于6号染色体和8号染色体的融合。但是关于癌细胞的染色体相互作用增加的信号是由于重组还是三维基因组组织的变异导致的还不清楚,因此针对这一问题,作者首先为“正常”的三维基因组组织特征建立了概率模型,包括位点、TADs、A/B compartments之间的基因组距离,发现小染色体和次端粒区域之间相互作用的增加。并且在重排的情况下,两个重排区域的基因是融合的,因其改变了位点之间的线性距离,从而也导致了与局部预期交互频率的偏差(图2a、b)。其次,作者利用Hi-C数据进行全基因组SVs检测,这一检测属于一种新型算法。该算法具体体现为:作者首先用一个特征良好的慢性粒细胞白血病细胞系(K562)来评估,并将结果与已发表的核型进行比较。在19个Hi-C预测的重排中,11个可以确认,其余8个是新的。由于这8个均在两个独立实验室进行的,它们不太可能是克隆进化的产物。随后,作者进行了FISH实验来验证新的预测易位。使用Hi-C数据预测的19个易位中有18个通过FISH或以前的核型验证,结果表明,新算法能够识别具有高特异性的大规模结构变异。
最后,将Hi-C分析扩展到27个癌细胞系和9个核型正常细胞系(图2d),发现在癌细胞中报告了25次重排,在正常细胞中几乎没有发生这种情况,染色体间和染色体内重排的比率约为2:1(所有细胞系中为424比274)。因此,新算法似乎可以识别大部分的大SVs,只有4.3%的无法识别。
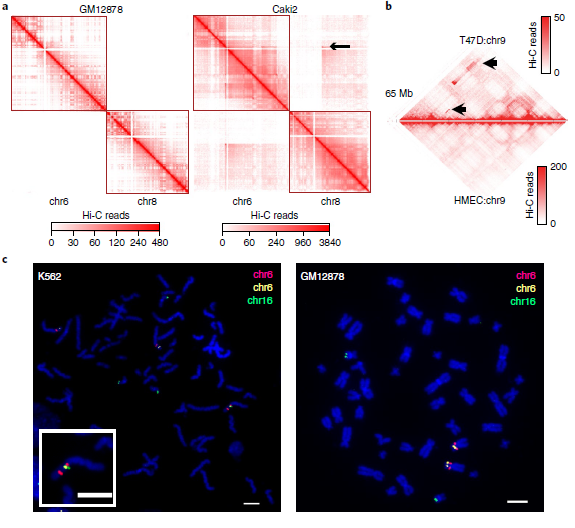
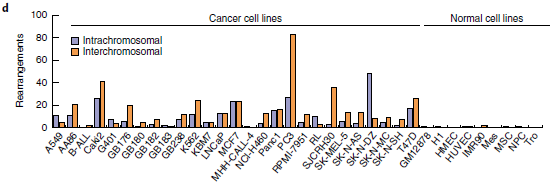
图2 利用Hi-C数据检测大规模重排
3. 不同方法检测SVs的比较
通过光学映射和WGS在每个癌细胞株中鉴定了数千个遗传物质的增加或损耗,光学映射检测到的缺失比WGS更少但范围更大。在T47D细胞中,WGS检测到2943个缺失,中位大小为552 bp,而Irys检测到1128个缺失,中位大小为1 335 bp(图3a,b)。其中85% WGS检测到的缺失被Irys遗漏,且其中78%的中位大小小于1kb。由于其分辨率受两个刻痕点之间的最小距离的限制,这些特征很可能被光学映射所忽略。Irys预测的缺失中有3%与多个较小的WGS缺失重叠,在这些情况下,这些WGS缺失的总和大小接近Irys检测到的缺失,但在Irys检测到的缺失中,有15%没有被WGS捕获。
作者测试了Irys检测到的一部分缺失,其中87.5%的缺失(16个缺失中的14个)通过了PCR验证。光学映射可以识别WGS reads没有被映射的重复区域内的缺失(图3c),以及在断点周围可映射性较低的区域。同时还发现WGS、Irys和Hi-C可以检测到不同染色体间大规模重排,类似基因组的非模板化添加碱基或外源DNA序列,如病毒的碱基,它可能来自第三条染色体因其太短而无法识别。如图3d所示,光学映射到局部结构,WGS用来确定断点,WGS通过定位断点和Hi-C数据来验证同一等位基因上几个相邻重排。总之,采用互补技术的综合方法对于更全面地了解癌症基因组的结构变化至关重要。
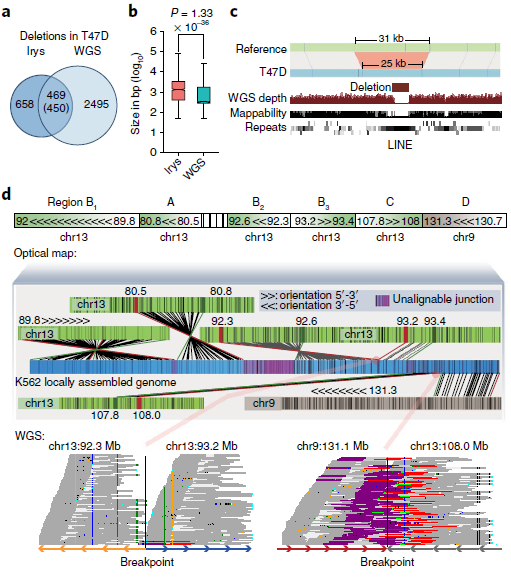
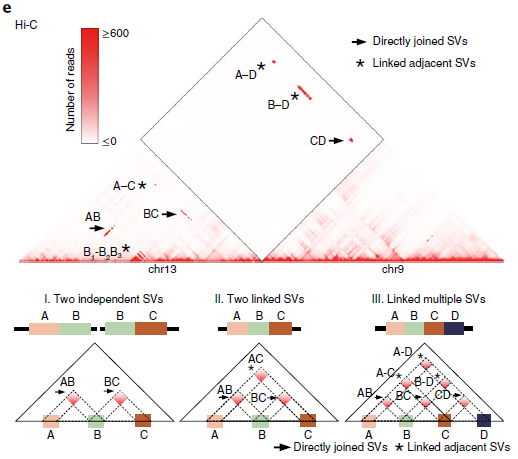
图3 不同方法检测SVs的比较
4. SVs对增强子的影响
拷贝数改变(CNAs)代表癌症的另一类遗传变异。作者在T47D乳腺癌细胞系中对CNAs进行了分析,并与560例乳腺癌患者的WGS数据进行了比较。在10个最常见的突变癌基因中,有8个在T47D癌细胞中被扩增,ATRX、CDKN1B等肿瘤抑制基因表达缺失(图4a),说明T47D细胞反映了乳腺癌的CNA表达情况。
作者进一步比较了T47D和人乳腺上皮细胞(HMECs)的RNA-seq数据,发现杂合性(LOH)缺失和纯合缺失导致基因表达显著降低,并且在其他癌细胞株中也观察到这一点。作者在25个COSMIC(癌症体细胞突变目录)发现与肿瘤相关的基因,大多数(76%)显示转录降低。而已知的癌基因(如MYC)和细胞周期检查点基因(如CDKN2A和CDKN2B)广泛扩增。
为了研究SVs是否可以通过破坏远端调控元件来影响癌症相关基因的表达。作者重点比较了T47D乳腺癌细胞与人乳腺上皮细胞(HMECs)。主要方法是使用ENCODE 联合 (URLs)的H3K27ac染色质免疫沉淀测序数据预测HMECs中的增强子,并将增强子与T47D中的缺失区域进行比较。结果表明,GNB4基因下游的3.4 kb缺失与乳腺组织特异性增强子重叠。由于基因组扩增,该区域有6个拷贝,其中5个带有这种缺失,只有单拷贝的增强子没有被破坏。
HMECs中的Hi-C数据表明GNB4可能受到单拷贝增强子的调控。更重要的是,它是该区域唯一表达减少的基因,该区域其余基因的表达高度上调,可能是由于拷贝数增加(图4c)。此外,发现缺失的增强子位于乳腺癌相关通路的基因附近(图4d),并且连接这些缺失的增强子相关的基因表达水平降低(图4e)。总的来说,这些结果表明癌症基因组的缺失可能经常影响增强子,并可能促进肿瘤发生。
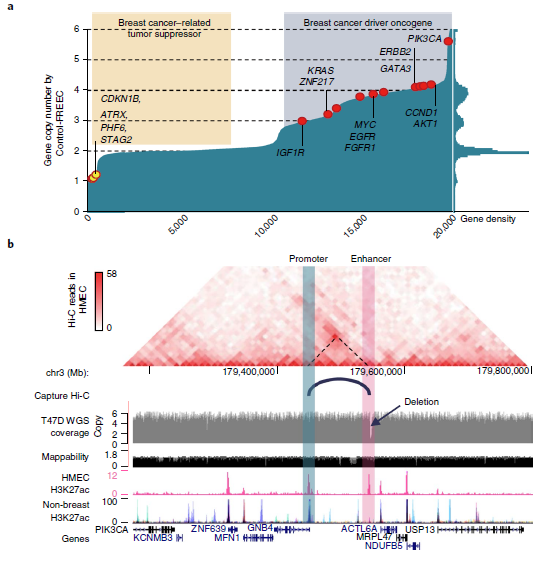
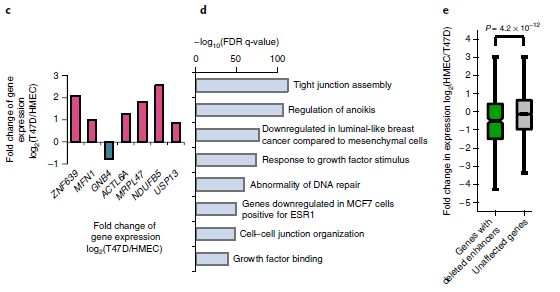
图4 SVs对增强子的影响
5. 结构变异对三维基因组组织的影响
基因突变会破坏拓扑结构域(TADs)并产生“新TADs(neo-TADs)”,导致发育障碍中的基因表达失调。为了研究SVs对三维基因组组织的影响,作者利用Hi-C数据鉴定了20个癌细胞株中的SVs,系统地研究了结构变化对TAD结构的影响,观察到neo-TADs是癌细胞大规模基因组重排的结果。如图5a所示,在PANC-1细胞中,染色体9和18的融合形成了一个neo-TAD。此外,发现许多由SVs诱导的neo-TADs在癌细胞中含有已知癌症驱动基因,如ERBB2、ETV1、ETV4、MYC、TERT等。
为了探讨neo-TAD形成是否是癌症基因组SV重排的一般结果,作者对每个细胞系中的所有断点交叉Hi-C信号进行了汇总分析。如图5b所示,观察到染色体间Hi-C信号形成一个尖锐的三角形(虚线),表明由于重排而形成的融合TAD。当使用随机边界位置打乱TADs进行同样的分析时,没有观察到这种情况。这些结果表明,癌症中的结构变异可以重组TAD结构,导致TAD融合和调节环境的改变(图5c)。
接着,作者研究了neo-TADs对基因表达的影响。在8个癌细胞系中,观察到包含重排的TADs基因比未重排的TADs基因表现出更大的等位基因偏差,这表明neo-TADs可能导致基因表达的改变。接着,作者检查了3个神经母细胞瘤细胞系的Hi-C数据,并比较了MYC的表达。其中SK-N-DZ具有较高的MYCN/N-myc表达(图5e),其余两个SK-N-SH和SK-N-AS具有较高的MYC/c-Myc表达。值得注意的是,在两个高MYC表达的神经母细胞瘤细胞系(SK-N-SH和SK-N-AS)中,发现了MYC基因附近存在易位。据癌症细胞系百科全书数据记载,这两个细胞系中没有MYC扩增,而作者观察了两种情况下包含MYC基因的neo-TADs的形成(图5f,g),表明neo-TADs的形成可能与MYC激活有关。综上所述,neo-TADs的产生是癌症基因组重组的结果。
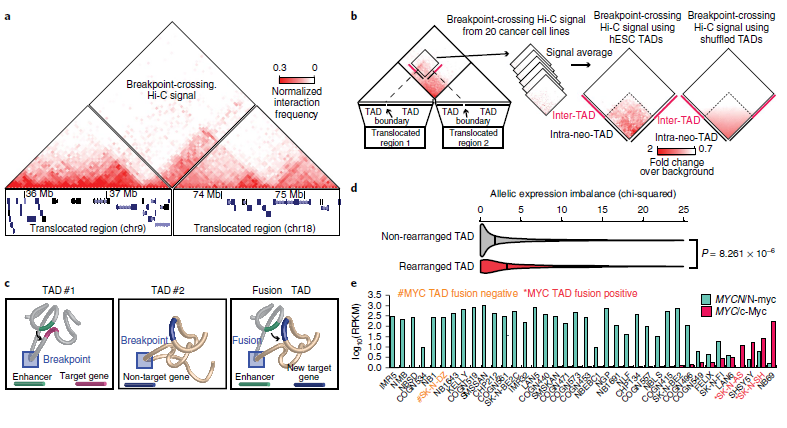
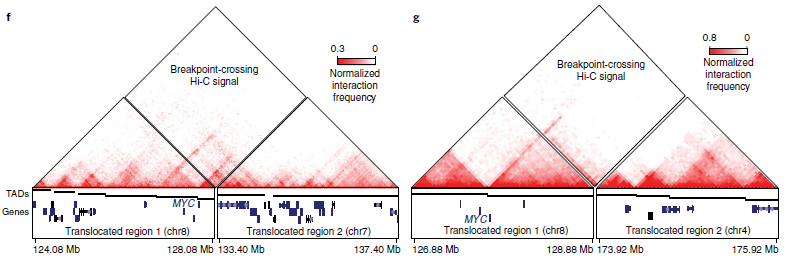
图5 重排和TAD融合
文章小结
作者利用高通量染色体构象捕获(Hi-C)、集成光学映射和全基因组测序,系统地检测正常或癌症样本中的SVs,发现光学映射和Hi-C能有效检测大而复杂的结构变化,高覆盖的WGS能识别高分辨率SVs。此外,还发现结构基因组变异而导致的三维(3D)基因组组织改变的实例,例如拓扑关联域(TADs)的形成或分解,这表明结构变异在肿瘤发生的基因失调中起着关键作用。
解析文献
Jesse R. Dixon, Jie Xu ,et al. Integrative detection and analysis of structural variation in cancer genomes. Nature Genetics, 2018, 50: 1388–1398.
参考文献
1. Northcott, P. A. et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature ,2014, 511, 428–434.
2. Dileep, V. et al. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res. 2015, 25, 1104–1113.
3. Haas, B. et al. STAR-Fusion: fast and accurate fusion transcript detection from RNA-Seq. Preprint at https://www.biorxiv.org/content/ early/2017/03/24/120295,2017.
4. Marchal, C. et al. Genome-wide analysis of replication timing by nextgeneration sequencing with E/L Repli-seq. Nat. Protoc.2018, 13, 819–839.
相关阅读